PathAI 是病理学(疾病研究)领域 AI 驱动的技术工具和服务的领先提供商。我们的平台旨在通过利用图像分割、图神经网络和多实例学习等现代机器学习方法,大幅提高复杂疾病诊断的准确性以及治疗效果的测量。

传统的手动病理学容易出现主观性和观察者变异性,这可能会对诊断和药物开发试验产生负面影响。在深入探讨我们如何使用 PyTorch 改进诊断工作流程之前,让我们首先阐述不使用机器学习的传统模拟病理学工作流程。
传统生物制药的工作原理
生物制药公司发现新型疗法或诊断方法有许多途径。其中一种途径严重依赖病理学切片的分析来回答各种问题:特定的细胞通讯途径是如何运作的?特定的疾病状态是否与特定蛋白质的存在或缺失相关?为什么某个临床试验中的特定药物对某些患者有效而对其他患者无效?患者预后与新型生物标志物之间是否存在关联?
为了帮助回答这些问题,生物制药公司依赖专业的病理学家来分析切片并帮助评估他们可能遇到的问题。
正如您可能想象的,需要经过专家委员会认证的病理学家才能做出准确的解释和诊断。在一项研究中,一份活检结果被交给 36 位不同的病理学家,结果是 18 种不同的诊断,严重程度从无需治疗到需要积极治疗不等。在困难的边缘病例中,病理学家也经常征求同事的意见。鉴于问题的复杂性,即使经过专家培训和协作,病理学家仍然难以做出正确诊断。这种潜在的差异可能决定一种药物是获批还是在临床试验中失败。
PathAI 如何利用机器学习助力药物开发
PathAI 开发机器学习模型,为药物开发研发提供洞察,为临床试验提供支持,并用于诊断。为此,PathAI 利用 PyTorch 进行切片级推理,采用多种方法,包括图神经网络 (GNN) 和多实例学习。在此语境中,“切片”指的是玻璃切片的全尺寸扫描图像,玻璃切片是两块玻璃之间夹着薄薄的组织切片,经过染色以显示各种细胞结构。PyTorch 使我们的团队能够使用这些不同的方法共享一个通用框架,该框架足够强大,可以在我们需要的所有条件下工作。PyTorch 的高级、命令式和 Pythonic 语法使我们能够快速原型化模型,然后在获得所需结果后将这些模型投入大规模应用。
在千兆字节图像上进行多实例学习
将机器学习应用于病理学的一个独特挑战是图像的巨大尺寸。这些数字切片的分辨率通常为 100,000 x 100,000 像素或更高,大小为千兆字节。将完整图像加载到 GPU 内存中并对其应用传统计算机视觉算法几乎是不可能完成的任务。对完整的切片图像(100k x 100k)进行标注也需要大量时间和资源,尤其是在标注者需要是领域专家(通过委员会认证的病理学家)的情况下。我们通常构建模型来预测患者切片上的图像级标签(例如是否存在癌症),而切片图像覆盖了整个图像中的几千个像素。癌变区域有时仅占整个切片的很小一部分,这使得机器学习问题类似于大海捞针。另一方面,某些问题(例如预测某些组织学生物标志物)需要聚合来自整个切片的信息,由于图像尺寸的原因,这又很困难。所有这些因素在将机器学习技术应用于病理学问题时都增加了显著的算法、计算和物流复杂性。
将图像分解成较小的补丁,学习补丁表示,然后汇集这些表示来预测图像级标签是解决此问题的一种方法,如下图所示。一种流行的做法称为多实例学习 (MIL)。每个补丁被视为一个“实例”,一组补丁构成一个“包”。单独的补丁表示汇集在一起以预测最终的包级标签。从算法上讲,包中的单个补丁实例不需要标签,因此允许我们以弱监督的方式学习包级标签。它们还使用置换不变池化函数,这使得预测与补丁的顺序无关,并允许高效地聚合信息。通常使用基于注意力的池化函数,这不仅允许高效聚合,还为包中的每个补丁提供注意力值。这些值表示相应补丁在预测中的重要性,可以可视化以更好地理解模型预测。这种可解释性元素对于推动这些模型在实际应用中的采用非常重要,我们使用诸如加性 MIL 模型之类的变体来实现这种空间可解释性。在计算上,MIL 模型避免了将神经网络应用于大图像尺寸的问题,因为补丁表示是独立于图像尺寸获得的。
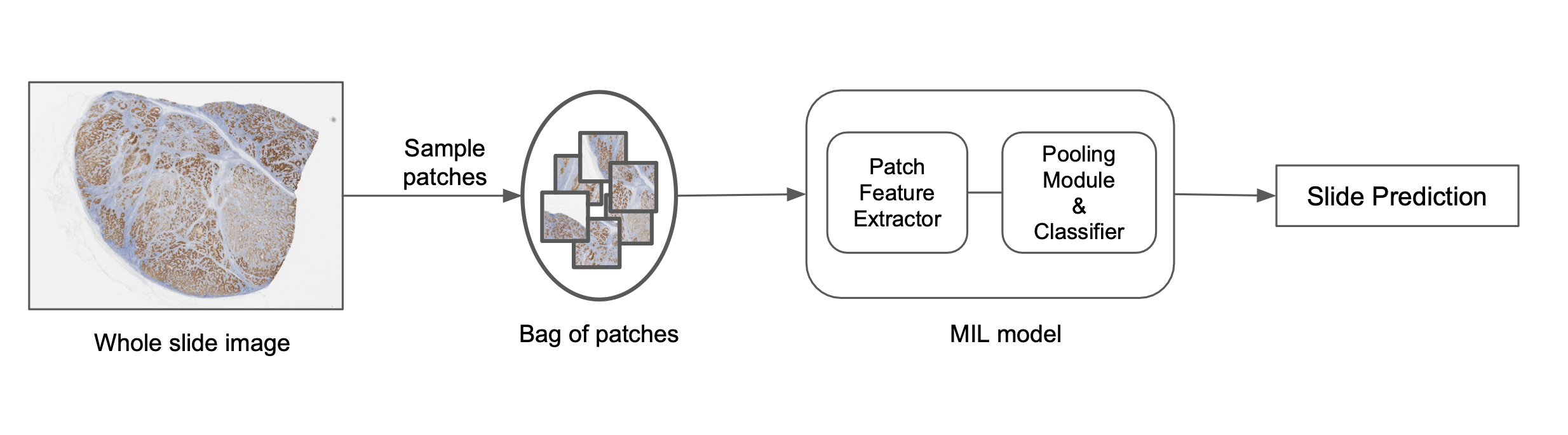
在 PathAI,我们使用基于深度网络的自定义 MIL 模型来预测图像级标签。此过程的概述如下:
- 使用不同的采样方法从切片中选择补丁。
- 根据随机采样或启发式规则构建补丁包。
- 根据预训练模型或大规模表示学习模型为每个实例生成补丁表示。
- 应用置换不变池化函数以获得最终的切片级分数。
现在我们已经介绍了 PyTorch 中 MIL 的一些高级细节,让我们看一些代码,看看使用 PyTorch 从构思到生产代码是多么简单。我们首先定义采样器、变换和 MIL 数据集。
# Create a bag sampler which randomly samples patches from a slide
bag_sampler = RandomBagSampler(bag_size=12)
# Setup the transformations
crop_transform = FlipRotateCenterCrop(use_flips=True)
# Create the dataset which loads patches for each bag
train_dataset = MILDataset(
bag_sampler=bag_sampler,
samples_loader=sample_loader,
transform=crop_transform,
)
定义了采样器和数据集之后,我们需要定义我们将用该数据集实际训练的模型。PyTorch 熟悉的模型定义语法使得这很容易实现,同时还允许我们创建定制模型。
classifier = DefaultPooledClassifier(hidden_dims=[256, 256], input_dims=1024, output_dims=1)
pooling = DefaultAttentionModule(
input_dims=1024,
hidden_dims=[256, 256],
output_activation=StableSoftmax()
)
# Define the model which is a composition of the featurizer, pooling module and a classifier
model = DefaultMILGraph(featurizer=ShuffleNetV2(), classifier=classifier, pooling = pooling)
由于这些模型是端到端训练的,它们提供了一种强大的方式,可以直接从千兆像素的全切片图像生成单个标签。由于它们对不同生物问题的广泛适用性,它们的实现和部署有两个重要方面:
- 对管道的每个部分(包括数据加载器、模型的模块化部分以及它们之间的交互)进行可配置的控制。
- 能够快速迭代构思-实现-实验-生产化循环。
PyTorch 在 MIL 建模方面具有多种优势。它提供了一种直观的方式来创建具有灵活控制流的动态计算图,这对于快速研究实验非常有用。Map-style 数据集、可配置的采样器和批处理采样器允许我们自定义补丁包的构建方式,从而加快实验速度。由于 MIL 模型是 IO 密集型的,数据并行和 Pythonic 数据加载器使任务非常高效和用户友好。最后,PyTorch 的面向对象特性使得构建可重用模块成为可能,这有助于快速实验、可维护的实现和轻松构建管道的组合组件。
使用 PyTorch 中的 GNN 探索空间组织结构
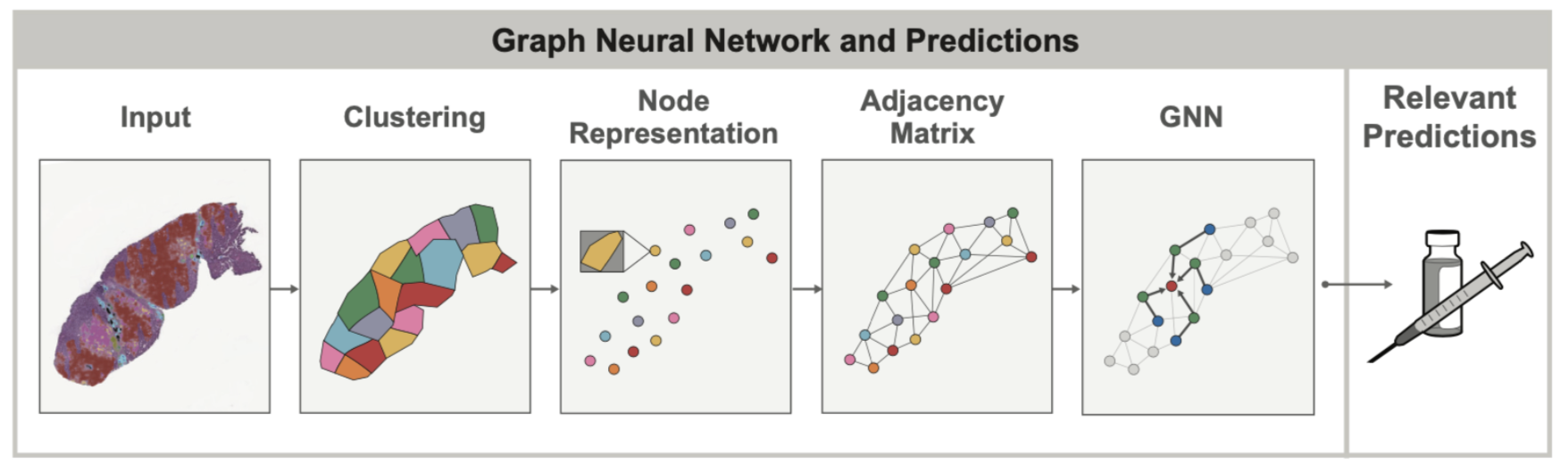
在健康和患病组织中,细胞的空间排列和结构通常与细胞本身一样重要。例如,在评估肺癌时,病理学家会观察肿瘤细胞的整体分组和结构(它们是否形成实心片状?还是以更小、局部的簇状存在?),以确定癌症是否属于具有截然不同预后的特定亚型。细胞与其他组织结构之间的这种空间关系可以使用图来建模,以同时捕获组织拓扑和细胞组成。图神经网络 (GNN) 允许学习这些图中与其他临床变量(例如某些癌症中基因的过度表达)相关的空间模式。
在 2020 年末,当 PathAI 开始在组织样本上使用 GNN 时,PyTorch 通过 PyG 包提供了最好、最成熟的 GNN 功能支持。这使得 PyTorch 成为我们团队的自然选择,因为 GNN 模型是我们知道将要探索的重要 ML 概念。
GNN 在组织样本背景下的主要附加价值之一是图本身可以揭示仅凭肉眼检查难以发现的空间关系。在我们最近的 AACR 出版物中,我们展示了通过使用 GNN,我们可以更好地理解肿瘤微环境中免疫细胞聚集体(特别是三级淋巴结构,或 TLS)的存在如何影响患者预后。在这种情况下,GNN 方法用于预测与 TLS 存在相关的基因表达,并识别超出 TLS 区域本身且与 TLS 相关的组织学特征。在没有机器学习模型辅助的情况下,从组织样本图像中识别此类基因表达的见解是很困难的。
我们成功应用的最有前途的 GNN 变体之一是自注意力图池化。让我们看看如何使用 PyTorch 和 PyG 定义我们的自注意力图池化 (SAGPool) 模型
class SAGPool(torch.nn.Module):
def __init__(self, ...):
super().__init__()
self.conv1 = GraphConv(in_features, hidden_features, aggr='mean')
self.convs = torch.nn.ModuleList()
self.pools = torch.nn.ModuleList()
self.convs.extend([GraphConv(hidden_features, hidden_features, aggr='mean') for i in range(num_layers - 1)])
self.pools.extend([SAGPooling(hidden_features, ratio, GNN=GraphConv, min_score=min_score) for i in range((num_layers) // 2)])
self.jump = JumpingKnowledge(mode='cat')
self.lin1 = Linear(num_layers * hidden_features, hidden_features)
self.lin2 = Linear(hidden_features, out_features)
self.out_activation = out_activation
self.dropout = dropout
在上面的代码中,我们首先定义一个卷积图层,然后添加两个模块列表层,这些层允许我们传入可变数量的层。然后我们取我们的空模块列表并追加可变数量的 `GraphConv` 层,然后是可变数量的 `SAGPooling` 层。我们通过添加JumpingKnowledge 层、两个线性层、我们的激活函数和我们的 dropout 值来完成 `SAGPool` 定义。PyTorch 直观的语法允许我们抽象出使用 SAG Poolings 等尖端方法的复杂性,同时保持我们熟悉的模型开发的通用方法。
像我们上面描述的 SAG Pool 模型这样的模型仅仅是 PyTorch 中的 GNN 如何让我们探索新颖想法的一个例子。我们最近还探索了多模态 CNN-GNN 混合模型,其准确率最终比传统病理学家共识分数高出 20%。这些创新以及传统 CNN 和 GNN 之间的相互作用再次得益于短期的研究到生产模型开发循环。
改善患者预后
为了实现我们通过 AI 驱动的病理学改善患者治疗效果的使命,PathAI 需要依赖一个机器学习开发框架,该框架能够 (1) 在开发和探索的初始阶段促进快速迭代和轻松扩展(即代码即模型配置)(2) 将模型训练和推理扩展到海量图像 (3) 轻松且稳健地为我们产品的生产用途(在临床试验及其他领域)提供模型服务。正如我们所展示的,PyTorch 为我们提供了所有这些功能以及更多功能。我们对 PyTorch 的未来感到无比兴奋,迫不及待地想看看我们还能使用该框架解决哪些其他具有影响力的挑战。